One-step Structure Prediction and Screening for Protein-Ligand Complexes using Multi-Task Geometric Deep Learning
Kelei He, Tiejun Dong, Jinhui Wu, Junfeng Zhang·August 21, 2024
Summary
LigPose is a groundbreaking AI method for drug development, using geometric deep learning to predict protein-ligand complex structures in a single step. This approach represents the ligand and protein pair as a graph, optimizing the complex's 3D structure directly. LigPose incorporates learning of binding strength and atomic interactions as auxiliary tasks, eliminating the need for docking tools. The model demonstrates superior performance in drug research tasks, suggesting a promising AI-based pipeline for drug development.
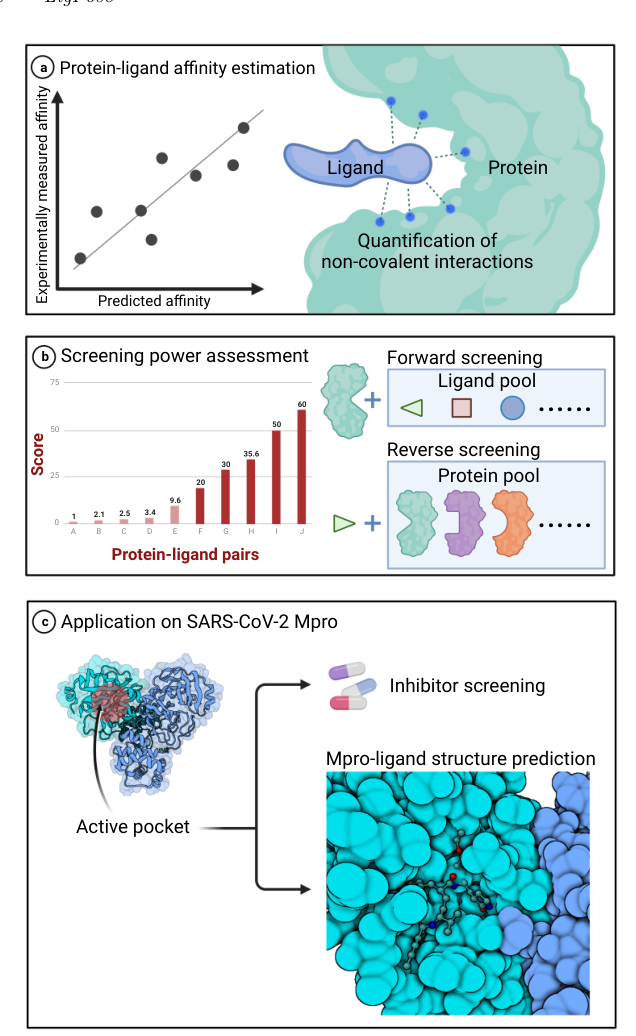
LigPose is a docking-free geometric deep learning method that provides refined atomic complex structures and screening for protein-ligand pairs. It uses a complete undirected graph to represent ligand and its binding target, optimizing 3D structures through atom coordinates in Euclidean space. LigPose excels in tasks like structure prediction, virtual screening, and affinity estimation, outperforming 12 existing docking tools by 14% in success rate for conformation prediction, with a 1851x faster inference speed. LigPose surpasses competitors by 20.1% and 19.3% in success rate for cross-docking and virtual screening, respectively.
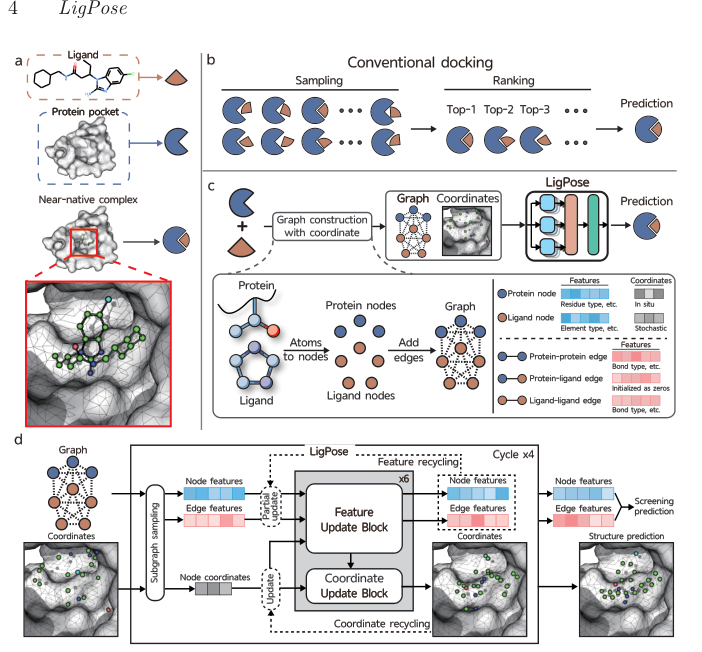
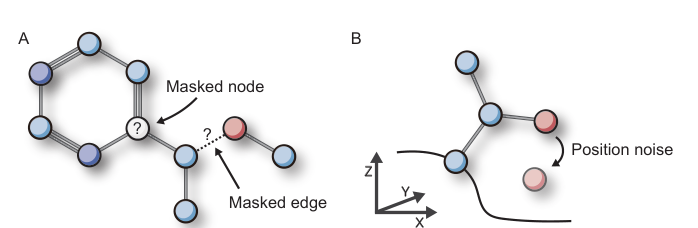
LigPose is a geometric deep learning-based method for predicting ligand-protein complex structures. It uses an end-to-end approach, contrasting with conventional docking methods. LigPose constructs graphs with protein and ligand nodes, and edges representing atom connections. It employs subgraph sampling, coordinate and feature recycling, and partial updates to predict structures. The method includes two prediction heads for auxiliary tasks and uses self-supervised learning on large-scale unlabeled data to improve generalizability. Experiments demonstrate LigPose's effectiveness in predicting flexible-ligand structures, comparing favorably with 12 popular docking tools on the PDBbind database's refined set.
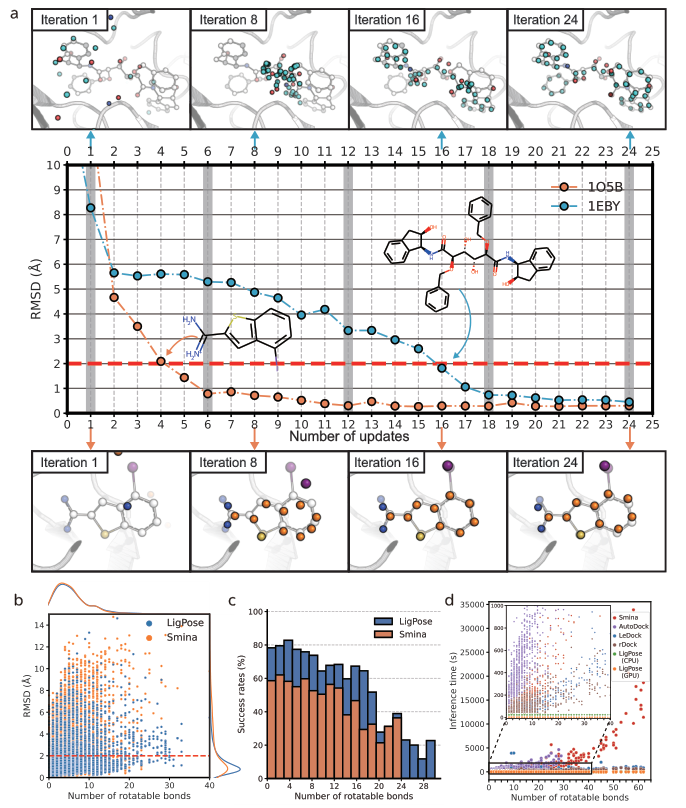
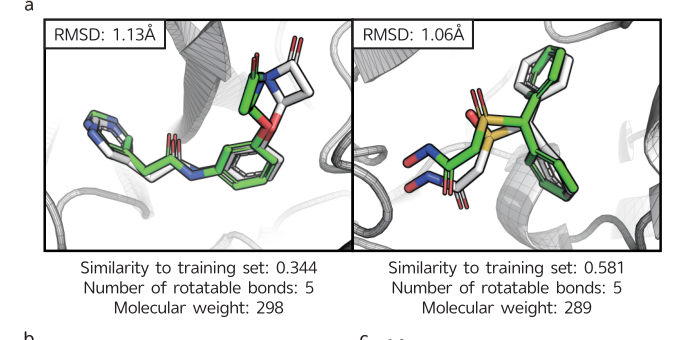
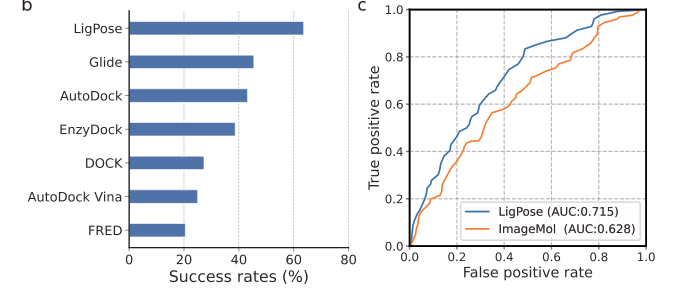
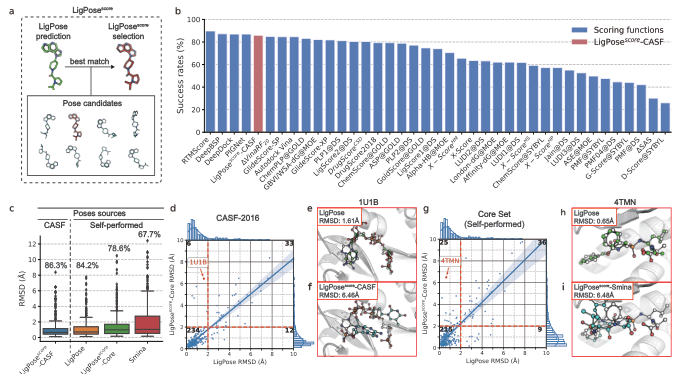
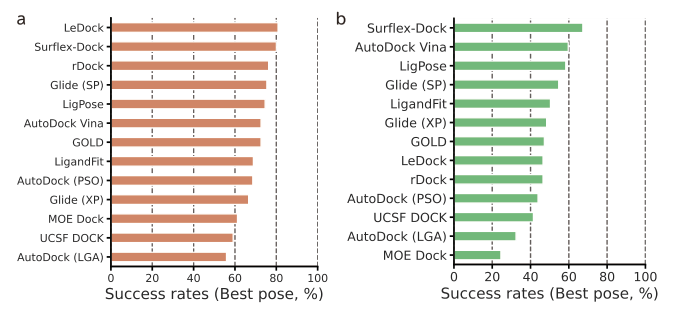
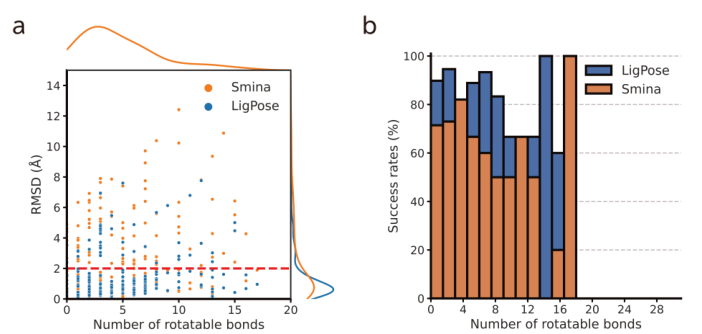
LigPose excels in predicting ligand poses within a benzene ring, achieving a success rate of 74.1% on a refined set, surpassing the best-performing docking tool, GOLD, by 13.5%. Other tools have lower success rates, ranging from 37.4% to 57.8%. Peptide-like molecules pose greater challenges due to their complex structures. LigPose surpasses 8 out of 10 docking tools when comparing poses with RMSD < 4Å for regular organic molecules. On the core set, LigPose outperforms the best-performing method, RTMScore, with a success rate of 84.2%. LigPose can also function as a scoring function, matching the performance of state-of-the-art scoring functions. In cross-docking, a more challenging task, LigPose achieves the highest success rate of 72.0%, significantly outperforming the second-best method, RTMScore, by 20.1%.
In conclusion, LigPose is a groundbreaking AI tool for drug development, using geometric deep learning to accurately predict ligand poses within protein targets and their respective binding strengths in one step. This method represents ligand and protein pairs as graphs, optimizing 3D structures directly. LigPose incorporates learning of binding strength and atomic interactions as auxiliary tasks, eliminating the need for docking tools. It demonstrates superior performance in drug research tasks, suggesting a promising AI-based pipeline for drug development.
Introduction
Background
Overview of AI in drug development
Importance of accurate protein-ligand complex structure prediction
Objective
Aim of LigPose in drug development
Key benefits of LigPose over traditional methods
Method
Graph Representation
Ligand and protein pair as a complete undirected graph
Atom coordinates in Euclidean space for structure optimization
Auxiliary Tasks
Learning of binding strength and atomic interactions
Elimination of the need for docking tools
End-to-End Approach
Contrast with conventional docking methods
Graph construction with protein and ligand nodes, atom connections as edges
Advanced Techniques
Subgraph sampling, coordinate and feature recycling, partial updates
Two prediction heads for auxiliary tasks
Self-Supervised Learning
Utilization of large-scale unlabeled data for improved generalizability
Performance
Comparative Analysis
Outperformance of 12 existing docking tools
Success rate in conformation prediction, virtual screening, and affinity estimation
Specific Tasks
Prediction of ligand poses within a benzene ring
Comparison with 10 docking tools for regular organic molecules
Success rate in cross-docking and virtual screening tasks
Applications
Drug Research Tasks
Structure prediction
Virtual screening
Affinity estimation
Comparison with State-of-the-Art
LigPose's performance against other scoring functions
Superiority in predicting ligand poses and binding strengths
Conclusion
Summary of LigPose's Contributions
Accurate prediction of ligand poses and binding strengths
Elimination of the need for docking tools
Integration of auxiliary tasks for enhanced performance
Future Directions
Potential for broader applications in drug development
Ongoing research and improvements in LigPose's capabilities
Basic info
papers
biomolecules
machine learning
artificial intelligence
Advanced features